 |
 |
|

|
|
|
|




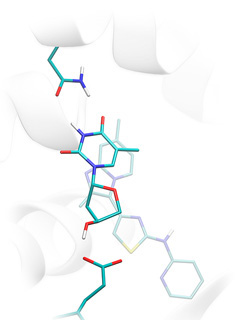 True active ligand of thymidine kinase correctly docked to the protein binding site and highly scored by Lead-Finder (opaque licorice) and low-scored inactive compound (semi-transparent licorice). |
Outstanding efficiency of ligand docking and binding energy estimations achieved by Lead-Finder are due to unique docking algorithm and extra precision representation of protein-ligand interactions. Docking algorithmFrom mathematical point of view ligand docking represents global minimum search on the multidimensional surface describing the free energy of protein-ligand binding. With ligands having up to 20-30 degrees of freedom (freely rotatable bonds) and complex nature of energy surface, global optimum search represents generally unsolved scientific task. To tackle this computationally challenging problem Lead-Finder applies unique approach combining genetic algorithm search, local optimization procedures, smart exploitation of the knowledge generated during the search run. Rational combination of different optimization strategies makes Lead-Finder efficient in terms of coarse sampling of ligand?s phase space and refinement of promising solutions. Entire docking algorithm has several tenths of settings, each of which has certain impact on the algorithm performance, robustness and speed of calculations. These relations were studied by us and for the sake of user comfort all settings were zipped in to two regimes of ligand docking algorithm with carefully balanced speed/accuracy ratio. First one is the default docking regime, which settings were adjusted to achieve maximum docking accuracy at a reasonable time of calculations. Second is the so-called screening regime, which was designed to be maximally fast at the cost of small (~5%) decrease in docking accuracy (see section Benchmarks). More details about the docking algorithm of Lead-Finder. Scoring FunctionExtra precise representation of protein-ligand interactions implemented in Lead-Finder scoring function is the second (in addition to docking algorithm) component of successful ligand docking. Lead-Finder scoring function is based on a semi-empiric molecular mechanical functional, which explicitly accounts for different types of molecular interactions. Individual energy contributions are scaled with empiric coefficients to fit particular purposes: accurate binding energy predictions, correct energy-ranking of docked ligand poses, correct rank-ordering of active and inactive compounds during virtual screening experiments. For these reasons three distinct types of scoring functions based on the same set of energy contributions but different sets of energy-scaling coefficients are used by Lead-Finder. First type of scoring function is designed for accurate estimation of free energy of ligand binding based on the structure of protein-ligand complex, and for this reason is called ΔG-scoring function. Experimentally determined structure of protein-ligand complex, or a modeled structure (obtained for example by docking or/and molecular dynamical studies) can sever as input for binding energy estimation with ΔG-scoring function. Scaling coefficients for this function were derived by fitting calculated binding energies to the experimental values for the set of protein-ligand complexes with known 3D-structure and experimentally measured binding constants (see section Accuracy of binding energy estimations). Second type — ranking scoring function — serves for correct ranking of ligand poses during the docking run. The purpose of this scoring function is to give highest score to the correct (experimentally observed) ligand pose. Parameterization (choice of scaling coefficients) of this scoring function aimed maximum docking success rate (maximum number of top-scored poses with correct geometry) for the fixed set of docking-predicted ligand poses over the set of protein-ligand complexes with known 3D-structures (see section Docking success rate). Finally, special set of scaling coefficients was designed to yield maximum efficiency in virtual screening experiments, that is — to assign higher score to active ligands (true binders) than to inactive ones. The latter scoring function type is called virtual screening or VS-scoring function. Assessment of VS-scoring function is illustrated in section Virtual Screening Performance. More details about Lead-Finder scoring function.
|
|
 |