| |
|
|
The quality of Lead-Finder predictions was extensively benchmarked in a series
of computational experiments:
- Accuracy of binding energy estimations
Ability of Lead-Finder to estimate free energy of protein-ligand
binding was benchmarked against the set of 330 diverse protein-ligand
complexes, which is currently the most extensive benchmarking study of such kind. Lead-Finder
demonstrated unique precision of binding energy prediction (RMSD = 1.5 kcal/mol)
combined with high speed of calculations
(less than one second per compound on average).
- Docking success rate
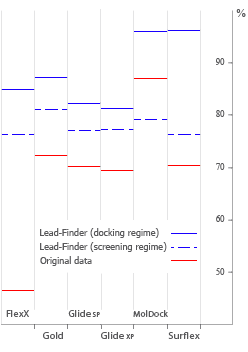
Accuracy of protein-ligand docking was assessed on the set of 407 structures,
which combines almost all published test sets of such programs as
FlexX,
Glide SP, Glide XP,
Gold,
LigandFit,
MolDock,
Surflex.
of Lead-Finder successfully predicted structures
of 85% of protein-ligand complexes, which is currently the best result
compared to other docking programs.
- Virtual screening performance
Ability of Lead-Finder
to distinguish active compounds from inactive during virtual screening experiments was benchmarked
against 34 therapeutically relevant protein targets. For almost all targets
Lead-Finder demonstrated impressive enrichment indicators, confirming its applicability
for in silico lead compound discovery.
- Speed of docking calculations
To estimate time and machine resources needed for your computational tasks, speed of ligand
docking on a single processor/core was benchmarked on different compounds
(with varying number of freely rotatable bonds), computer architectures, settings
of the docking algorithm.

| |