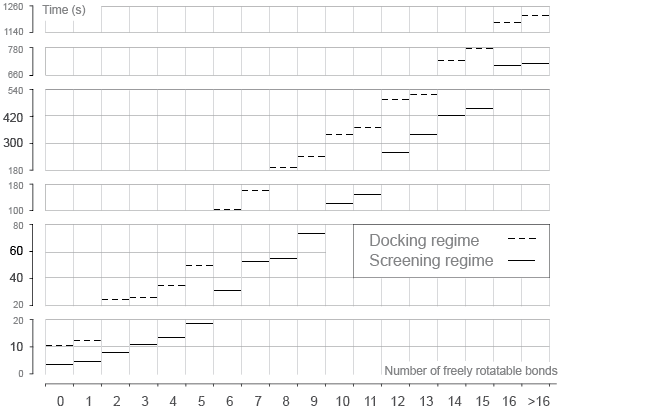
Speed of ligand docking calculations depends on the settings of genetic algorithm search.
Currently two types of settings are predefined in Lead-Finder: more accurate and exhaustive
search in the default docking regime and faster search in the screening regime.
While default docking settings were adjusted to yield maximum docking accuracy, settings for
the screening regime were ´lightened‘ to achieve maximum speed of calculations at the cost of small (~5%) decrease in Docking Success Rate.
To benchmark speed of docking calculations a set of 407 diverse protein-ligand
complexes (used also for Docking Success Rate measurements) was used. All calculations were performed
in two regimes: default docking and screening. Results of current benchmarking studies providing
average time needed to dock ligand with a certain degrees of freedom (number of freely
rotatable bonds) are presented on the figure. As can be seen from the figure, screening
regime performs 2–4 times faster than default docking, taking on average 30 seconds for
docking ligand with 6 freely rotatable bonds.
 |