 |
 |
|

|
|



|
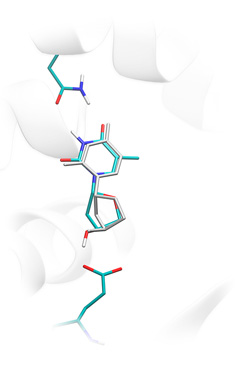
Ligand binding to thymidine kinase: crystallographic position (grey licorice) and position predicted by Lead-Finder (colored licorice). You can download examples of protein-ligand docking using Lead-Finder. Each example (archive file) contains description of the task and necessary structure files.
1dm2: CDK2 complex with inhibitor hymenialdisine — marine sponge constituent.
|
Ligand dockingLead-Finder predicts the structure of a protein-ligand complex by docking a flexible ligand to a static protein structure. The outstanding efficiency of Lead-Finder in finding the correct geometry of protein-ligand complexes is based on a unique conformational search algorithm, and an extra precision scoring function to evaluate the free energy of ligand binding (see the Technology section). Lead-Finder can dock ligands with up to 20–30 degrees of freedom; non-covalent and covalently bound protein-ligand complexes, as well as ligand binding to metalloproteins can be treated equally robust. Five- and six-member rings of the ligand are treated flexibly during docking. Accuracy of protein-ligand docking was validated on the set of 407 protein-ligand complexes, which is currently the most extensive benchmarking study of such kind. Our test set was composed of test sets of such docking programs as FlexX, Glide SP, Glide XP, Gold, LigandFit, MolDock, Surflex, which allowed straightforward comparison of Lead-Finder and original results for the competitive programs. As can be seen from our benchmarking studies, Lead-Finder outperformed all competitive programs on their native test sets, with docking success rate ranging from 80.0% on Glide XP and FlexX test sets to 96.0% on Surflex and MolDock test sets. Lead-Finder has intuitive program interface, which is easy to configure and use. Though the entire docking algorithm has several tenths of settings, all of them, for the sake of user comfort, were zipped in to two regimes with carefully balanced speed/accuracy ratio. First one is the default docking regime, which settings were adjusted to achieve maximum docking accuracy at a reasonable time of calculations. Second is the so-called screening regime, which was designed to be maximally fast (2-4 times faster than the default docking regime) at the cost of small (~5%) decrease in docking accuracy (see Benchmarks section). Time needed to dock one ligand on a single processor/core using Lead-Finder depends primarily on the number of freely rotatable bonds of the ligand and volume of the protein binding site. Speed of ligand docking calculations was extensively benchmarked by us, results can be found in the corresponding section. To characterize briefly Lead-Finder performance, during our computational experiments on a cluster of 16 computational nodes (dual Xeon 5150 2.6 GHz) computers we were able to screen 300000 of compounds (STK library by Vitas-M Laboratory) in 12-24 hours (depending on the protein target) in the screening regime.  |
|
 |