 |
 |
|

|
|



|
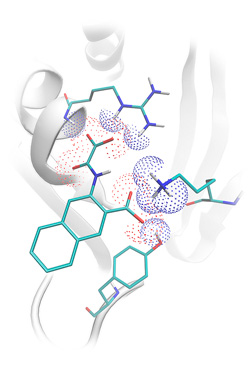
Position of protein tyrosine phosphatase inhibitor predicted by Lead-Finder (reference pdb structure 1c84).
Electrostatic and hydrogen bonding interactions are presented by dot surface.
You can download examples of protein-ligand binding energy estimations using Lead-Finder. Each example (archive file) contains description of the task and necessary structure files.
1hvr: HIV-protease complex with nonpeptide cyclic urea inhibitor.
|
Binding energy calculationsLead-Finder software can be used for extra precision calculations of the free energy of protein-ligand binding. This functionality is quite unique to docking software, since binding energy calculations are usually included in separate software packages that are based on such theoretical approaches as free energy perturbation or linear interaction energy. These packages require a high level of expertise in computational chemistry, and are often time and investment intensive. Thanks to the original semi-empiric molecular mechanical scoring function implemented in Lead-Finder, extra precision calculations of the free energy of ligand-protein binding can be performed on the flight with ligand docking. No extra computational/expert time is required, and neither is any investment in additional software. The precision of binding energy estimation with Lead-Finder was validated on a set of 330 experimentally characterized protein-ligand complexes with a broad distribution of ligand binding affinities and other physicochemical properties (see Benchmarks section). This yielded a RMSD of 1.5 kcal/mol from experimental data. Binding energy calculations can be performed concurrently with ligand docking, or for a predetermined protein-ligand structure (obtained from experimental data or other molecular modeling studies). Ability of Lead-Finder to accurately predict free energy of ligand binding is hard to overestimate in drug discovery studies, modeling ADMET properties in silico, studies of enzyme’s specificity and rational enzyme design.  |
|
 |